
- Proteomics 3단계
- 특정 세포에서 발현되는 모든 단백질을 분리
- Protein mapping
- 각 spot의 단백질을 분석한다. 단백질 염기서열이 DNA처럼 잘 발달되지 않았기 때문
- PMF (Peptide Mass Fingerprinting)
- 아미노산 서열이 아닌, 단백질을 잘라서 펩타이드 개수와 질량을 분석
- MALDI-TOF 기법을 이용한 질량분석기로 측정
- PST (Peptide Sequence Tag)
- 펩타이드 말단에서 떨어져 나간 아미노산을 질량분석으로 결정
- PMF (Peptide Mass Fingerprinting)
- 각 단백질을 규명한다.
- MALDI-TOF MS
- ESI-MS
- 특정 세포에서 발현되는 모든 단백질을 분리
- MALDI-TOF
- Mass spectrometry (질량 분석기)
- 전기장 하에서 전하를 띤 물체는 힘을 받아 운동하며, 운동의 크기는 물체의 질량과 전하의 비에 따라 다르다.
- TOF(Time of Flight) : 전기장 하에 움직인 거리를 측정하여 질량을 알아낸다.
- 질량이 작을수록, 전하량이 클수록 이동시간이 빠르다.
- MALDI : 매트릭스를 섞어서 레이져를 쏴서 이온화
- 과정
- 트립신(Trypsin)으로 자른다.
- 잘린 펩타이드를 이온화시킨다. (이온화 보조제 = MATRIX)
- 분자량 작은 펩타이드가 먼저 도착한다.
- 도착 시작을 이용하여 분자량을 역으로 추정한다.
- 데이터베이스에 기록된 자료와 비교한다.
- Yeast two-hybrid **
- 단백질을 모르고 DNA만 갖고 있을 때, 두 유전자의 산물이 서로 상호작용을 하는가를 알아보는 방법
- 각각의 단백질을 융합시켰을 때 상호작용을 한다면, 직접 붙어있었던 것처럼 전사활동을 해서 유전자를 발현시킬 수 있다.
- 전사 인자에 단백질을 붙여 전사가 나타나는가를 관찰하며 단백질 상호작용 여부를 판단
- Activation Domain과 Binding Domain이 있다.
단백질체학에서의 연구 대상은 전체 단백질이다.
그래서 proteome이라고 한다.
단백질 수준에서 왜 연구해야하냐?
1. mRNA 에서 서열이 번형돼서 가공되면 단백질에서도 그럴 수 있으므로 아미노산 서열도 달라질 수 있다.
2. mRNA 양만을 가지고 최종적인 단백질 양과 일치하는지는 알 수 없다.
3. 단백질의 변형을 mRNA분석을 통해 알기 힘들다.

가장 중심이 되는 분석 기법 :
특정세포에서 만들어진 모든 단백질을 하나하나 규명하는 것 3단계
1. 특정세포에서 발현되는 모든 단백질을 분리
Gel 상에서 전개 해논다.
2. 각 spot에 있는 단백질들을 오려서 단백질이 뭔지를 본다.
(1) 아미노산을 결정하는 것이 가장 좋지만 DNA염기서열처럼 잘 발달되진 않았다.
Peptide mass fingerprinting (PMF)
단백질을 잘라서 펩타이드 개수와 질량을 분석한다.
(2) Peptide Sequence Tag (PST)
펩타이드의 염기서열의 꼬리를 결정한다.
3. database에 있는 것으로 PMF와 PST를 알 수 있다.
(1) MALDI-TOF
(2) ESI-MS
Mass spectrometry

단백질을 잘라서 나온 펩타이드 조각의 질량을 분석하는 것
전기 장에 놓으면 + - 극으로 이동하게 된다.
운동량은 질량의 크기와 전하량과 관련이 있을 것이다.
이동시간은 질량이 작을수록 전하량이 클수록 빨리 간다.
질량 대 전하비 = 질량/전하 의 값이 클수록 늦게 도달하게 된다.
질량이 작으면 더 빨리 도달하고 질량이 크면 더 늦게 도달한다.
전기장 내에서 움직인 거리나 비행시간(TOF)로부터 질량을 알아낼 수 있다.
MALDI-TOF의 이온화 과정과 전기장에서의 움직임 측정 2단계
- 이온화 : 매트릭스 보조를 시킨 다음에 레이저를 쏴서 이온화를 시킨것이 MALDI 이다.
매트릭스를 섞어서 레이져를 쏴서 이온화 시킨 것 - TOF 측정 : 이동한 시간을 잰다.
질량이 적을수록 빨라진다.

질량이 작으면 빨리 도달.

단백질의 경우 2차원으로 한다.
아래쪽으로 분자량에 따라서 분리를 하면 분자량에 따라 자기 위치에 가 있다.
이것만 했을 때는 다른 종류의 단백질이 뭉쳐있다.
2차원으로 하면 다 전개가 된다.

하나는 분자량에 따라서, 또 하나는 pH에 따라서 분류한다.

이 경우에서는 1차적으로 pH gradient로 분류한다.
이걸 가지고 분자량에 따라 다시 전개함으로써 분류한다.
그리하여 의미가 있다고 판단되는 반점에서 단백질을 추출하였다. 그러면 이 단백질이 어떤 단백질인지 어떻게 알 수 있는가?
각 스팟의 단백질을 뽑아서 어떤 유전자로부터 발현된 단백질인지를 알고싶다.
맨 처음에 할 것은 트립신(Trypsin)으로 자르는 것이다.
트립신은 Arg, Lys을 자른다.
그 후 트립신으로 잘린 펩타이드들의 분자량을 측정해야 한다.
말디-토프 질량분석기(MALDI-TOF MS)를 이용하여 펩티드의 분자량을 측정한다.
Matrix Assisted Laser Desorption/Ionization Time Of Flight (MALDI-TOF)

펩티드를 고출력 레이저를 이용하여 (1)이온화시킨다.
그리고 관에 전기장을 걸어주면 이온화된 펩티드가 전기장에 의해 관을 이동하는데, (2)분자량이 작은 녀석이 먼저 도착한다.
(3)도착한 시간을 이용하면 분자량을 역으로 추정할 수 있다. 전기영동과 원리 자체는 거의 같다. 단지 펩티드를 이온화시키는 단계가 추가된 것 뿐이다.
트립신으로 처리한 펩티드는 단백질에 따라 고유한 분자량을 가지므로, 단백질 (4)데이터베이스에 기록된 자료와 비교하면 이 펩티드가 어떤 단백질인지 알아낼 수 있다.

(b) mass가 작은쪽이 더 빨리 도달해서 빨리 검출이 된다.
이 단백질을 트립신으로 자르면 5개의 펩타이드가 나왔다.
이 펩타이드의 개수와 peak의 위치는 특정단백질 고유의 지문과 같다.
(a) 이온화 시키는 것
이온화 보조제 = MATRIX
전기장을 걸면 이동하게 되고 질량이 작고 전하량이 큰 것이 먼저 도달하게 돼서
(b)에서 각 피크들의 위치를 얻을 수 있다.
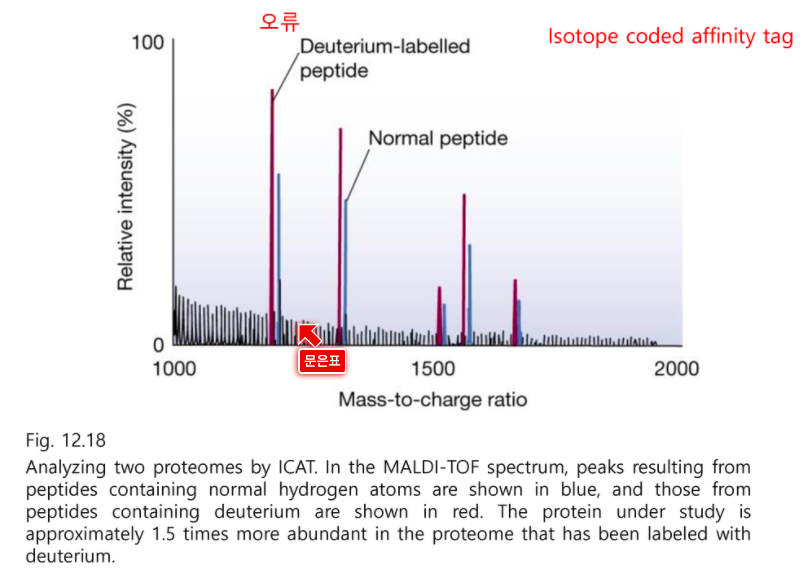
오류가 있다. 특정단백질의 발현량이 있다.
하나의 세포는 그냥 수소로 표지하고 다른 것은 중수소로 표지했다.
피크를 비교해보면 오른쪽인 normal한 펩타이드보다 중수소로 표지한 펩타이드가 더 무거워야한다.

이 기법이 바로 Isotope coded affinity tag 이다.

우연히 Mass finger print가 유사할 수 있다
PST는 petide 의 끝 아미노산 서열을 비교하는 것이다.
3단계를 연속적으로 수행해야한다.
방이 3개가 있다.
각각의 펩타이드를 얻어서 일정한 곳으로 가게 해서 TOF에 따라 분리하고 mass를 결정한다.
여기서 통과돼서 나오는 것을 두번째 방에서는 불활성기체로 충돌시킨다.
그러면 아미노산들이 떨어진다.
남아있는 펩타이드의 분자량을 결정한다.
세번째 방에서는 남아있는 펩타이드 분자량의 질량을 분석한다.


3번째 방에서 질량을 결정한다.

트립신은 아르기닌과 라이신을 자른다.
이것을 가지고 peptide mass fingerprint를 결정한다.


펩타이드가 우연히 일치할 수 있다.
그래서 세번째 방에서 아미노산 몇개가 날라가고 남은 fingerprint를 결정하면 날아간 아미노산을 유추할 수 있다.

Y6 -> y5 : 하나 날라간 펩타이드
L이라는 아미노산의 차이가 질량의 차이이다 17
'🧬 Bio > 유전공학' 카테고리의 다른 글
| 유전공학 13장 (1) (0) | 2020.05.25 |
|---|---|
| 유전공학 12장 (3) (0) | 2020.05.21 |
| 유전공학 12장 - Studying Genome (0) | 2020.05.07 |
| 유전공학 11장 - Studying gene expression and function (0) | 2020.05.04 |
| 유전공학 10장 - Next Generation Sequencing (2) (0) | 2020.04.27 |