
3.1.6 세균 이외의 생물체로부터의 total cell DNA의 분리
실험 목적상 세균 이외의 생물체로부터 total cell DNA를 분리할 필요가 있다.
물론 세균에서의 경우와 동일하지 않으며, 차이점은 다음과 같다.
1) 세포 파괴 단계에서 차이점
식물 세포 : lysozyme이 효과가 없기 때문에 물리적 방법(냉동시킨 시료를 갈아서 부수는 등)을 사용하거나, cellulase[셀룰로스(식물 세포벽의 구성성분) 분해효소]를 사용하여 세포벽 파괴
동물 세포 : 세포벽이 없기 때문에 detergent 처리만으로 쉽게 용해시킬 수 있음.
2) 세포 추출물 내용물의 차이
세균 : 주로 단백질, DNA, RNA
식물 : 단백질, DNA, RNA 외에 다량의 탄수화물(광합성의 결과) 존재
구조가 다르기 때문에 식물에서 라이소자임은 필요가 없다.
그런데 식물에서는 액체 질소에서 동결해서(얼려서) 막자사발에서 갈면 식물세포가 깨진다.

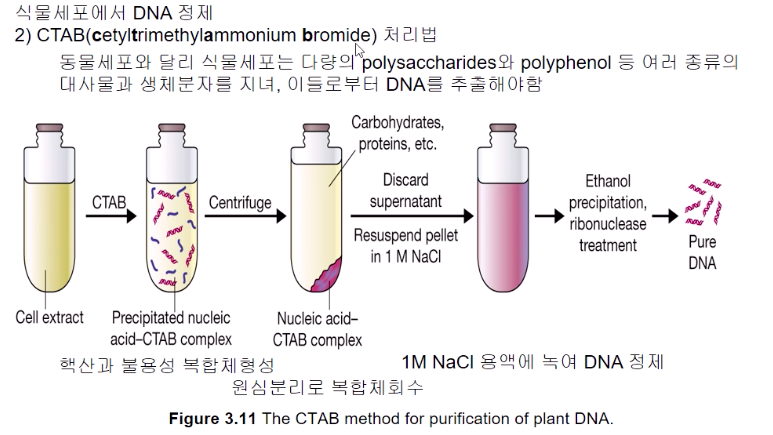
CTAB을 처리하면 핵산의 침전을 형성해서 원심분리를 하면 잘 가라앉는다.
불용성복합체를 형성한다.
1M NaCl에 녹이면 CTAB과 DNA가 분리된다.
이걸 물에 녹인 다음에 RNAse를 넣고 에탄올에 또 넣으면 pure한 DNA를 얻을 수 있다.
동물은 굉장히 쉽다.
지금까지 동물, 식물, 박테리아로부터 DNA를 추출하는 방법을 배웠다.
3.2 플라스미드 DNA의 분리
Total cell DNA 분리 후 염색체 DNA와 플라스미드 DNA를 분리시킨다.
* 염색체 DNA와 플라스미드 DNA의 차이점 이용
크기 : 가장 큰 플라스미드의 크기는 대장균 염색체 크기의 8%밖에 안됨
conformation : 분자 전체의 공간적인 configuration. 플라스미드와 세균 DNA는 모두 circular form이지만 분리도중 세균 DNA는 linear form으로 절단된다. 이 차이로 인해 alkaline denaturation, EtBr-CsCl 밀도구배 원심분리(density gradient centrifugation)를 통해 분리 가능

그러면 이제부터 벡터를 준비해야한다.
벡터에는 플라스미드와 박테리오페이즈가 있다는 걸 배웠다.
이제부터는 박테리아에서 플라스미드를 분리하는 법과 박테리오페이즈를 분리하는 법을 배워보자.
플라스미드는 굉장히 다양하다고 했다.
플라스미드는 크기에 따라서 또 다르다.
3.2.1 크기에 기초한 분리

조심스럽게 세포를 용해시킨다면 세균 DNA의 단편화가 감소하게 되므로 원심분리에 의해 쉽게 플라스미드 DNA를 분리할 수 있다.
1. lysozyme과 EDTA , 그리고 25% 수크로스(sucrose)처리하여 세포를 스패로플라스트[sphaeroplast(partially wall-less cells)]로 만듦.
2. sphaeroplast의 세포막을 파괴하기 위해 Triton X-100(non-ionic detergent)처리
3. 세포 추출물을 원심분리(10,000 rpm, 5~10 min.)
4. 세포 찌꺼기는 침전, 상층액의 상단엔 cleared lysate(플라스미드 포함)가, 하단엔 큰 DNA 단편들이 존재
5. 그러나 짧게 단편화된 염색체 DNA가 플라스미드와 함께 존재할 수 있으며 플라스미드가 큰 경우에는 세포 찌꺼기와 함께 침전되었을 수도 있다. 즉, 크기에 의해선 완전히 분리할 수 없다.
다른 찌꺼기도 있지만 박테리아의 염색체도 사이즈가 크다.
박테리아 염색체가 세포막에 붙어있으면,
오염물질로부터 큰 사이즈의 플라스미드를 박테리아의 염색체와 분리하는데 용이하다.
아까와 같이 라이소자임과 EDTA를 넣어서 세포벽을 적절하게 파괴시킨다.
아직 세포막은 남겨논다.
그리고 25퍼센트 sucrose를 넣는다.
세포막의 삼투압을 유지시켜줄 수 있다.
세포막이 파괴되는 것을 일단 잠시 멈춰논다. 등장액처럼
그 다음 여전히 세포막에 붙어있는 채로 있는다.
Triton X-100은 mild 한 detergent이다.
여전히 염색체가 박테리아의 세포막에 붙어있게 한 채로 세포막을 깨뜨린다.
SDS 같은 것은 박테리아의 염색체를 파손시킬 수 있다.
그래서 Triton X-100은 파손시키지 않고 분리할 수 있다.
비이온성이다.
염색체를 파손되지 않게, 잘리지 않게 세포막을 깬다.
염색체가 굉장히 크기때문에 가라앉지만 플라스미드는 둥둥 뜨게 된다.
과정 요약
- 세포벽 깬다 (세포막 안깨지게) (EDTA + Lysosome)(with Sucrose)
- 세포막 깬다 (염색체 파손 안 되게) (Triton X-100)
- 원심분리
- 상층액 추출(플라스미드 DNA 추출)
3.2.2 구조(conformation)에 따른 분리

그래서 그 다음 또 하는 방법은 구조에 따라서 하는 방법이다.
똑같은 circular DNA라 하더라도 supercoil이나 Open-circular이 생기면서 수퍼코일이 사라지는 게 있다.
DNA정제 과정에서 염색체 DNA는 일부는 파괴된다.
그러나 플라스미드는 supercoil 을 유지한다.
박테리아의 염색체는 절대로 supercoil을 유지할 수 없게 된다.
플라스미드 DNA 는 supercoil을 유지할 수 있다.
그래서 supercoil만 정제하면 플라스미드를 빼낼 수 있다.
이 방법으로 플라스미드만 정제하는 방법은 alkaline denaturation이라 한다.
(a) Alkaline Denaturation

* 대부분의 플라스미드 : supercoiled
(a) Alkaline denaturation : 극한 pH 변화 조건에서 non-supercoiled DNA는 변성되고 supercoiled plasmid는 변성되지 않는 성질을 이용한 방법임.
main chromosome의 linear dsDNA와 supercoiled plasmid의 혼합액을 pH 12.0~12.5로 만든다.
그러면 linear dsDNA는 변성되어 ssDNA가 된다.
이 혼합액을 pH 7.0으로 만든다.(use 3M NaOAc(pH 4.5))
그러면 linear ssDNA는 무작위적인 수소결합을 형성함으로써 서로 엉키면서 거대한 덩어리를 이루게 된다.
원심분리하면 상층액에는 supercoiled plasmid가 존재하며 이때 생기는 pellet은 바로 linear DNA이다.
플라스미드는 수퍼코일을 유지하게 된다.
수퍼코일을 유지한 플라스미드와 linear상태의 염색체를
알카라인으로 pH를 확 높여주면 linear 한것들은 이중가닥이 전부 떨어져서 single-strand로 분리가 된다.
그 후 pH를 서서히 낮춰서 다시 7.0으로 낮추면, 박테리아의 염색체는 엉켜버리게 된다.
이걸 원심분리하면 엉켜버린것들은 밑으로 가라앉고 supercoiled 된 것들은 상층액에 남게 된다.
따라서 알카라인으로 분리하게 되면 페놀이나 ribonulcease를 할 필요가 없다.
단백질과 RNA가 함께 제거되기 때문이다.

NaOH -> 선형 DNA의 이중가닥을 단일가닥으로
acetic acid -> pH를 낮추면서 박테리아 DNA는 tangled 된다.
(b) Ethidium bromide-caesium chloride density gradient centrifugation
(EtBr-CsCl 밀도구배 원심분리)

Ethidium bromide-caesium chloride density gradient centrifugation(EtBr-CsCl 밀도구배 원심분리)
: plasmid를 가장 순수하게 분리할 수 있는 방법
CsCl density gradient centrifugation에서, 고속(40,000 rpm)의 원심분리 안(48hr) 원심력은 Cs과 Cl ion을 튜브(tube)의 바닥쪽으로 당기기 때문에 농도구배가 형성되어 CsCl 밀도는 바닥쪽으로 갈수록 더 커진다. 고분자와 함께 원심분리되면 분자의 고유한 밀도와 동일한 CsCl밀도 위치에 밴드(band)를 형성하게 되며 DNA는 1.7g/㎤, 단백질은 상단부, 그리고 RNA는 바닥에 pellet을 형성하게 된다.
EtBr과 함께 density gradient centrifugation이 되면 non-supercoiled DNA와 supercoiled DNA를 분리시킬 수 있다. EtBr은 DNA의 염기쌍 사이에 삽입됨으로써 linear DNA는 0.124g/㎤만큼, supercoiled DNA는 0.085g/㎤만큼 밀도를 감소시킨다(부력은 증가). 결과적으로 supercoiled DNA는 EtBr-CsCl gradient에서 linear와 oc(open circular)DNA와는 다른 위치에 band가 형성된다.
세포 추출물과 함께 EtBr, CsCl를 첨가하고 40,000 rpm 으로 48시간 원심분리시킨다. CsCl 밀도구배가 형성되며, EtBr에 의해 부력의 차이가 생긴 linear & ocDNA는 상단에, supercoiled DNA는 하단에 band를 형성하게 된다.(최상단엔 단백질, 최하단 pellet에는 RNA가 존재)
cf>EtBr : DNA 염기 사이에 끼어 들어감으로써 부력 변화시킴
linear and ocDNA : 0.125g/㎤ 밀도 감소
supercoiled DNA : 0.085g/㎤ 밀도 감소
supercoiled DNA 의 band만을 주사기로 추출하여 새로운 tube에 옮긴다. DNA 염기사이에 삽입되어 있는 EtBr을 제거하기 위해 n-butanol(n-부탄올)처리 반투막 이용하여 투석시킴으로써 CsCl과 DNA를 분리
구조에 따라서 플라스미드를 구분할 때 Ethidium bromide와 CsCl(세슘클로라이드)를 이용한다.
아래로 갈수록 CsCl의 농도가 높다.
부력을 통해서 플라스미드를 추출할 수 있다.
초고속원심분리를 통해 CsCl의 농도기울기를 형성한다.
그러면 부력밀도에 따라서 각각이 거기에 딱 고정된다.
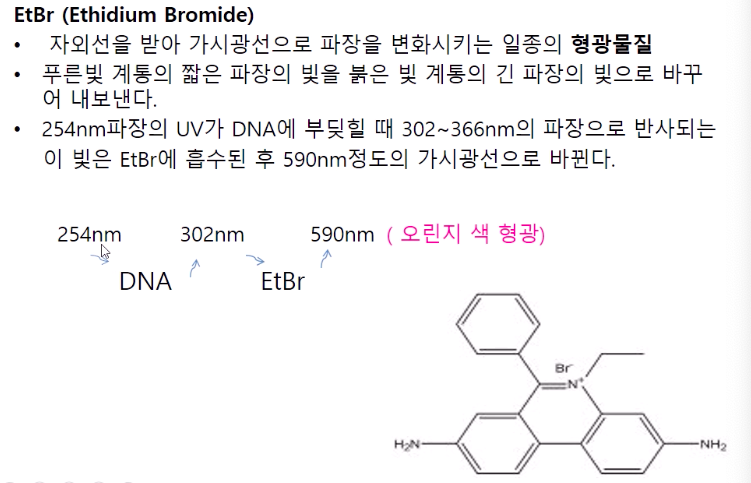
자외선을 쪼면 형광빛이 나타나게 되는걸 이용해서 DNA의 위치를 확인 할 수 있다.

보면 한 염기쌍과 염기쌍은 3.4 옹스트롬이고
한 헬릭스에는 10개정도의 염기쌍이 있다.
EtBr을 처리하면 염기쌍 사이사이에 삽입이 된다.
intercalate가 된다.
EtBr이 끼어들게 되면 부력밀도가 낮아지게 된다.
선형과 원형 DNA에서 이런 부력 밀도의 감소가 크게 차이가 나게 된다.
정상적일 때는 DNA의 밀도가 1.70g/cm^3 이다.
선형DNA 밀도가 더 많이 감소된다.
이 감소 차이를 이용해서 supercoiled 플라스미드를 잘 분리해 낼 수 있다.
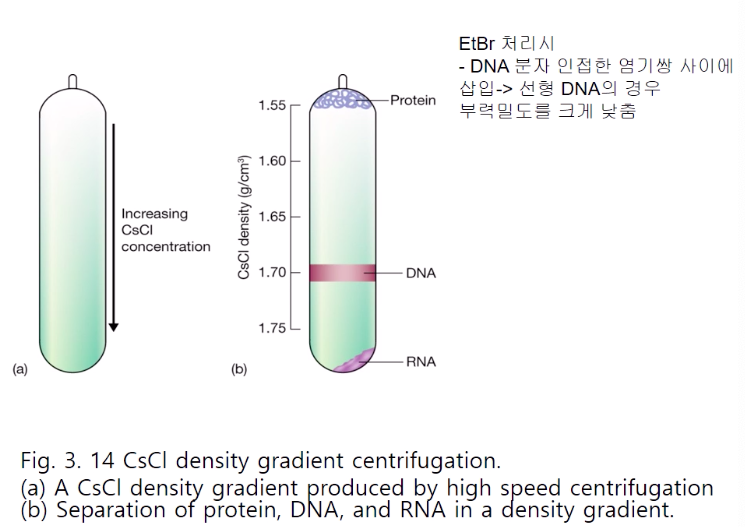
(a) 위에다가 샘플을 집어 넣는다.
그러면 각각의 부력밀도에 따라서 각자 위치로 간다.
그 후 UV를 쪼이면

DNA밴드의 위치를 확인할 수 있다.
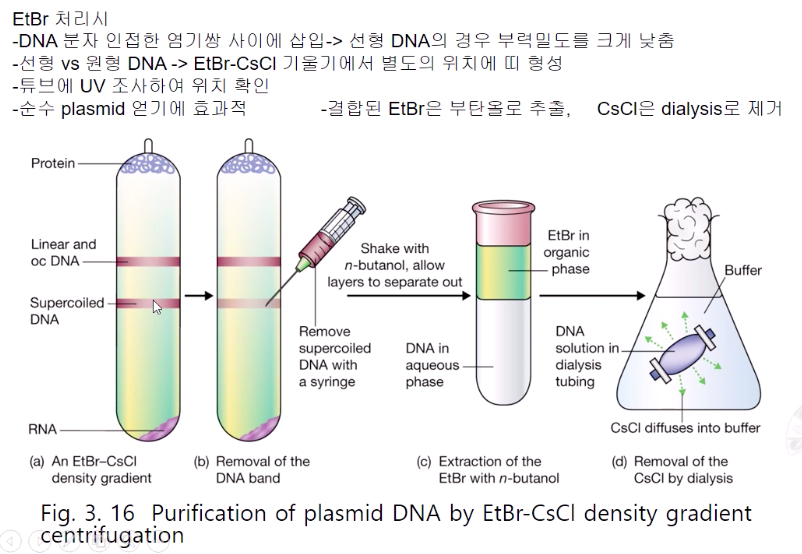
linear DNA와 supercoiled DNA의 밀도차이를 통한 위치 차이를 확인할 수 있다.
그러면 주사바늘로 supercoiled DNA를 빼내면 된다.
이 에띠디움브로마이드가 삽입된 상태인 DNA를 추출하게 된다.
이 에띠디움브로마이드를 DNA로부터 빼내야 한다.
이때 normal-부타놀을 사용한다.
튜브에 넣고 섞으면 수용액층이 밑에 오고 부타놀 층이 위로 뜨게 된다.
부타놀 층의 에띠디움브로마이드가 층에 가둬지게 된다.
상층액을 버리고 밑을 취한다.
dialysis : 세슘의 높아서 투석하듯이 세슘크로라이드를 빠져나가게 한다.
그러면 DNA만 모을 수 있다.
이렇게 구조의 차이로 벡터의 supercoiled 플라스미드를 추출할 수 있게 된다.
1. 크기에 따른
2. 구조에 따른 (1) alkaline denaturation (2) EtBr-CsCl gradient

헬릭스 턴을 26도 정도 풀어준다.
그러면 한 턴에 36개까지 들어갈 수 있게 된다. (정상은 10개가 한 턴)
트위스트 값이 감소되고 writhing값이 증가가 된다. (수퍼코일 넘버가 증가)
양의 방향으로 수퍼코일이 생기게 된다.
linking 넘버는 고정되어 있는데 twist값은 감소하게 되어서 supercoiled 넘버는 증가한다.


그래서 같은 DNA이지만 gel상에서 이동속도가 달라지게 된다.
똑같은 DNA도 supercoiled가 가장 이동속도가 빠르다.
구조에 따라서 gel상에서 이동속도가 달라지게 된다.
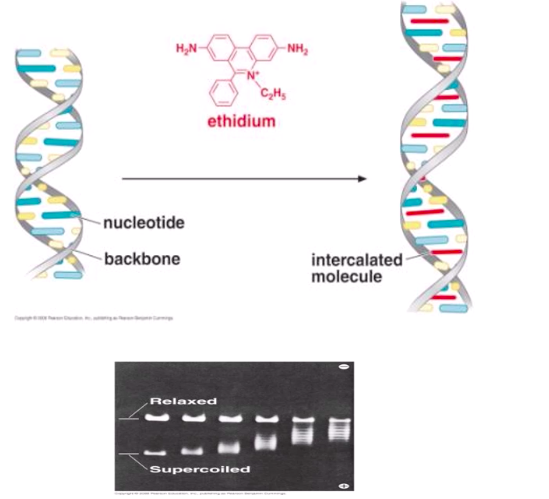
하나씩 하나씩 supercoiled 풀면 더 느려지는걸 볼 수 있다.

3.2.3 플라스미드 증폭

early log phase인 A600=0.3 일 때 단백질 합성 저해제(chloramphenicol;클로람페니콜)첨가.
; 12시간 더 배양 → 플라스미드 계속 복제
플라스미드를 최대한 늘려서, 하나의 세포가 엄청 많은 플라스미드를 갖게해야 한다.
플라스미드의 replication을 증가시킨다.
chloramphenicol을 처리하면 플라스미드만 복제가 가능하다.

단백질의 합성이 없어도 플라스미드 복제가 가능하다.

단백질 합성이 세균에서 억제가 된다.
그러면 세균의 염색체는 복제가 되지만
단백질 합성이 가능하지 않아도 플라스미드의 복제가 가능하게 된다.
세균의 자기 염색체의 복제가 일어나지 않지만, 플라스미드는 단백질의 합성과 상관 없기 때문에 복제가 계속 일어나게 된다.
이 replication inhibitor의 합성이 중지 돼서 copy number의 조절을 받지 않게 된다.
그래서 이러한 원리로 플라스미드의 복제만 가능하게 된다.
3.3 박테리오파지 DNA의 분리
다른 DNA분리와 가장 큰 차이점은 출발 물질이 세포 추출물이 아니라는 점이다.
왜냐하면 감염된 세균 배양액에서 세포밖 배지에 파지가 존재하기 때문이며 배양액을 원심분리시켜 상층액을 취한 후 캡시드를 제거하면 된다.

이걸 배우면 어떠한 대상에서 total DNA를 뽑아서
플라스미드나 박테리오페이즈에 삽입해서 클로닝하는 과정을 위해서
DNA를 프랩하는 방법을 배우게 된다.
지금까지 Cell로부터 total DNA를 뽑는 것은 insert를 준비하는 것
그 다음 벡터를 준비하는 것에서 플라스미드를 준비하는 법을 배웠고
이제 벡터 DNA에서 박테리오페이즈 DNA를 준비하는 법을 배운다.
박테리오페이즈에 감염된 세균 배양액에서 박테리오페이즈 입자를 회수한다.
원심분리를 통해 페이즈 입자를 회수한다.
이렇게 하면 titre가 낮다. (농도)
3.3.1 높은 λ 농도(titre or titer)를 얻기 위해 배양물을 증식시키는 방법

lysogenic상태에서는 너무 얻기 힘들다.

#Phage titre : meaning the number of phage particles per ㎖ of culture.
박테리오파지 λ는 용원성 사이클을 가지고 있기 때문에 세포외 λ titre는 매우 낮다.
따라서 용균성 사이클을 유도시켜야 한다.
그래서 cI 유전자[통합(integration)에 관여]이 불활성화된 ts 돌연변이체(mutant)(integration 후 30℃에서는 유도(induction)가 되지 않아 정상적으로 세포가 분열하지만, 42℃에서는 induction이 일어나 excision이 일어남으로써 용균성 사이클로 전환된다)를 숙주로 이용하여 용균성 사이클을 진행시킴.
ci 라는 유전자가 필수적으로 필요하다
ci 유전자의 돌연변이를 만들었다.
온도에 민감해서 42도로 높이면 프로파지의 상태로 존재하지 않는다.
lytic cycle로 가서 많은 입자를 얻을 수 있다.
그래서 적당히 키우다가 lytic cycle로 가게해서 얻는다.
돌연변이가 작용해서 프로파지상태로 있지 못하고 lytic cycle로 가게 된다.
3.3.2 비용원성(non-lysogenic) λ 파지의 제조

cI 유전자와 통합에 관여하는 다른 유전자들을 결손(deletion)시킨 λ를 이용한다.
이러한 λ는 용원성 사이클을 거치지 않고 바로 용균성 사이클을 진행시키게 된다.
이 파지로 높은 titre를 얻기 위해선 배양액을 증식시키는 정도가 중요하다.
: 숙주와 파지 밀도(농도)를 적절히 조절
①배양물 밀도가 너무 낮을 때 - 모든 세포가 빠르게 용해(용균)됨 → 낮은 파지 titre
②배양물 밀도가 너무 높을 때 - 배양물이 완전히 용해 안됨 → 낮은 파지 titre
③배양물 밀도가 적당할 때 - 배양물이 계속 증식하지만 결국에는 용해됨 → 높은 파지 titre
처음부터 ci유전자를 삭제한 걸 넣어준다.
그래서 감염을 시켜주면 대부분의 세균이 잘 감염돼서 lytic cycle로 가게 된다.
ci유전자를 삭제한 람다 페이즈를 사용한다.
그러나 들어가자마자 용균성을 잃게 되면 세균이 자라지도 못했는데 다 깨져버리게 된다.
들어가자마자 세균들이 죽어버리게 되고 증폭도 안되게 된다.
그래서 세균을 감염시킬 때 세균의 농도가 얼마인지가 굉장히 중요하게 작용한다.
여전히 박테리오 페이즈의 입자가 높지 않다.
적절하게 대부분의 세균이 감염되게 한 후 파티클을 얻게 된다.
세균은 적당히 크면서 감염이 계속 일어나게 되면서
용균성이 되면서 많은 파티클들을 얻게 된다.
궁극적으로는 세균이 감염이 되지만 용균성이 돼서 maximum의 파티클들을 얻게 된다.
3.3.3 감염된 배양물로부터 파지의 수집

세균 세포와 파지는 원심분리를 통해 구분이 가능하다.
그러나 파지는 크기가 작기 때문에 오랜 시간 원심분리해야 하는 불편함이 있다.
파지를 쉽게 농축시키기 위해 PEG를 이용한다.
PEG는 긴 사슬 구조의 중합 화합물(long-chain polymeric compound)로 수분를 흡수하여 고분자를 뭉치게 함으로써 파지를 침전시킨다.
침전된 파지를 원심분리 후 작은 부피로 재용해시킨다.
박테리아는 가라앉고 그 위에 페이즈 입자들을 모은 다음에 PEG라는 것을 첨가한다.
NaCl이 존재하면서 물을 흡수하면서 입자를 침전시킨다.
그래서 페이즈 파티클들이 침전되게 한다.
상층액을 버리고 다른 버퍼에 잘 녹인다. resuspend
그다음 껍데기를 벗긴다.
3.3.4 λ 파지 입자로부터 DNA의 분리

CsCl density gradient centrifugation 이용한다. λ 파지는 1.45~1.50 g/㎤에 band가 형성된다.
①숙주 세포 배양(ts mutant)
②λ 파지를 감염시킴
③원심분리 → λ 파지 분리
④PEG 처리 → λ 파지 농축
⑤CsCl density gradient centrifugation → λ 파지 분리
⑥투석 → CsCl 제거
⑦페놀 처리 → 단백질 제거
⑧EtOH → DNA 침전
박테리아의 염색체를 없에기 위해서
세슘크로라이드 밀도를 만들어 놓고
이 위에서 PEG침전물을 녹여서 로딩을 하고 초고속으로 원심분리를 한다.
그러면 단백질은 위에뜨고 RNA는 가라앉고 람다 페이즈 DNA만 추출할 수 있게 된다.
3.3.5 M13 DNA의 분리

숙주세포를 파괴시키지 않고 계속 자손 파지를 방출시키기 때문에 λ에 비해 높은 titre를 별다른 조치 없이도 쉽게 얻을수 있으며, 파지 현탁액이 세포 찌꺼기에 오염되지 않으므로 CsCl density gradient centrifugation 할 필요가 없다.
M13 DNA는 프로파지 상태로 존재하지 않기 때문에 많은 타이틀을 얻을 수 있다.

①숙주 세포 배양
②원심분리 : M13 파지 분리
③PEG + NaCl : 파지 농축, 원심분리
④완충액(buffer;버퍼)에 재현탁
⑤페놀 : 단백질 제거
⑥EtOH : DNA 침전
(a) 상층액만 얻는다. 박테리아를 버린.
(b) PEG 넣는다
(c) 상층액 버린다.
(d) 버퍼에 잘 녹인다
(e) 페놀 extraction 하면 캡시드 단백질 나온다
(f) M13 DNA만 에탄올 집어넣고 precipitation 시키고
(g) 잘 녹이면 M13 DNA만 준비가 된다.

이 준비된 insert DNA와 벡터 DNA를 사용해서
recombinant DNA를 만드는 것을 배워보자
'🧬 Bio > 유전공학' 카테고리의 다른 글
| 유전공학 4장 - Electrophoresis (0) | 2020.03.26 |
|---|---|
| 유전공학 4장 - DNA manipulation (0) | 2020.03.26 |
| 유전공학 3장 (1) - 세포로부터 DNA 분리, 정제 (0) | 2020.03.19 |
| 유전공학 2장 - Vehicles for gene cloning (0) | 2020.03.16 |
| 유전공학 1주 1회 (0) | 2020.03.16 |