scRNA-seq을 하는 2가지 이유
Bulk RNA-seq 분석이 불가능하므로
Bulk RNA-seq의 조직에 2가지 이상의 세포가 섞여서 전사체들이 bias를 만든다.
scRNA-seq은 세포끼리 섞이지 않기 때문이 bias가 없다.
초기 scRNA의 프로토콜
- 피펫으로 난자 캡쳐
- Cell lysis
- Reverse Transcription cDNA 합성
- Poly A tailing
- PCR
따라서 조직에서 세포를 하나씩 캡쳐하는 기술이 필요하다.
Barcode
CB (Cell Barcode) : 각각의 세포 구분
UMI (Unique Molecular Identifier) : 각각의 분자 구분 -> 증폭의 noise를 줄일 수 있다.
여러가지 캡쳐 방법이 존재한다.
- 많은 종류의 세포를 얻을 수 있는 시스템 = Large scale : 10 x
- splicing 이나 variant는 볼 수 없다.
- 관심있는 유전자를 집중적으로 보고싶을 경우 = Mid-to-low scale : FACS or SMART-seq
scRNA-seq의 도전 과제
- 단일세포 캡처 기술
- 증폭시 생기는 noise 컨트롤
Noise 생기는 2가지 원인
1. stochastic mRNA loss
*stochastic = random
capture efficiency가 세포의 종류와 유전자에 따라 다르기 때문이다.
2. 증폭 (amplification)
샘플 RNA의 양이 많을수록 노이즈를 감소시킬 수 있다.
Amplification factor가 세포 및 유전자 별로 다르다. -> Unique Molecular Identifier로 해결 가능
Single-cell Multi-omics
한 세포에서 RNA-seq만 하는 것이 아니라, DNA-seq, Epigenetics, Proteomics를 동시해 시도해볼 수 있다.
세포 lysis 후 핵만 뽑아 DNA-seq 하고, Cytosol에서 RNA-seq 이런식으로
이것을 Large-scale로 하는 것이 가장 중요한 이슈이다.
scRNA-seq 문제에 대한 CS적 해결책
High technical noise : 통계로 해결
High biological noise : (유전자 발현은 stochastic하여 bimodal : 봉우리가 두개)수학적 모델링으로 해결
Big data : (데이터가 많다) 기계학습을 통해 해결
scRNA-seq의 고유 특징
- Large-scale platform
- 10 x
- dorp-seq-sparse count matrix
- Count matrix에 0이 많은 이유
- 유전자 발현이 stochastic하기 때문에 발현 안됐을 수도 있다.
- Capture efficiency와 Sequencing depth가 낮기 때문에
- 많은 세포를 한번에 분석할 수 있다.
- condition을 모르는 경우가 많다. -> unsupervised 분석 수행
- clustering
- PCA
- t-SNE
- 반면 bulk RNA-seq은 input/output condition을 알고 있으므로 -> supervised 분석 수행
- DEG
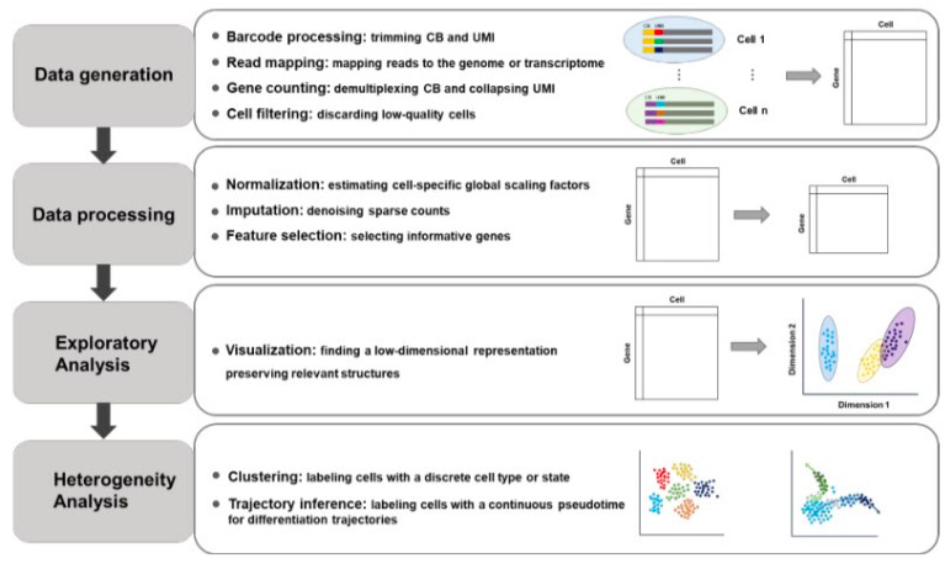
scRNA-seq의 workflow
- 데이터 생성 (Data generation) : 바코드 처리, read mapping, gene counting, Cell filtering
- 데이터 처리 (Data processing) : normalizaion, imputation, feature section
- 탐색적 분석 (Exploratory analysis) : 시각화 (visualization)
- 이종성 분석 (Heterogeneity analysis) : clustering, trajectory inference
아직 Gold-standard tool은 존재하지 않는다.
R과 Python 플랫폼이 가장 tool로써 많이 사용된다.
1. 데이터 생성 (Data generation)
- 바코드 처리 : 시퀀싱 기계가 생성한 read file(FASTQ)의 header에는 Cell Barcode(CB)와 UMI가 있다.
- Read mapping : Bulk RNA-seq와 동일하게 수행된다. 완료시 raw UMI count table 생성
- Gene counting : CB의 demultiplexing 및 UMI 합친다.
- Cell filtering : low-quality 세포를 제거해야 한다.
10x 에서 제공하는 프로그램 Cell Ranger Analysis Pipeline이 위 단계를 자동으로 분석해준다.
Cell filtering 1
- Read마다 header에 CB 및 UMI 정보가 있기 때문에, 각 read가 어디에 mapping이 되었나 정보가 나온다. 이걸로 UMI count table을 만들 수 있다.
- Count table을 만들 때 염두사항
- barcode를 도입함으로써 모든게 해결되는 것이 아니다 - sequecing error 존재
- PCR error
- 실제 데이터를 보면 seq-error와 PCR error 때문에 UMI의 종류가 늘어나는 것을 볼 수 있다.
2. 데이터 처리 (Data processing) - normalization, imputation, feature selection
위 분석을 수행하여 QC가 끝난 Gene X Cell count matrix를 얻는다.
실제 분석을 하기 전에 normalization, imputation, informative feature selection을 해야한다.
Normalization : 세포 특이적 global scaling factor를 예측한다.
Normalization을 하는 이유는 Gene X Cell count matrix의 Cell마다 bias가 들어있기 때문이다.
그러므로 bias를 없애서 각각의 gene이 cell마다 실제 발현값을 예측하는 것이 목적이다.
Normalization은 어떤 세포의 예상되는 유전자의 숫자가 유전자의 실제 발현 및 세포 특이적 global scaling factor와 비례한다는 것이 가정한다.
global scaling facotr = 세포크기, capture and RT efficiency, amplification factor, dilution factor, sequencing depth 등
R의 Scran package로 Normalization 이 가능하다.
3. 탐색적 분석 (Exploratory Analysis) - 시각화 (Visualization)
위의 informative gene을 사용하여 각각의 세포를 축소된 공간으로 projection 하기 = Dimentionally reduction
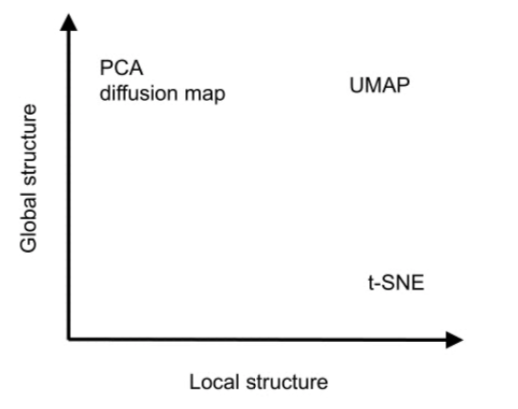
다양한 기법 : PCA, diffusion map, UMAP, t-SNE
축소된 공간에서 어떤 feature를 강조할 것인가 살펴봐야 한다.
local structure : 이웃 지역의 세포들 간 세포와 세포의 거리 유지
global structure : 생물학적 과정(cell cycle 등)과 관련된 저차원에서 세포와 세포 간 거리 유지
각 기법마다 강조하는 것이 다르다.
PCA, diffusion map : global structure 강조
t-SNA : local structure 강조
UMAP : local/global structure의 균형
4. 이종성 분석 (Heterogeneity Analysis) - clustering, trajectory inference
- Discrent latent variable 방법 : 세포의 종류와 상태를 나타내기 위한 clustering 방법
- Continuous latent variable 방법 : 각 세포가 어떤 생물학적 과정(ex 분화)에 속해 있나 나타내기 위한 방법
'🧬 Bio > 생명정보학' 카테고리의 다른 글
| contig 란? (0) | 2021.03.23 |
|---|---|
| IGV (Integrative Genomics Viewer) (0) | 2021.02.01 |
| Read count (0) | 2021.01.20 |
| RPKM, FPKM, TPM (0) | 2020.11.15 |